Workflow for Identification of Differentially expressed genes using RNAseq data
This automated pipeline assists in identification of differentially expressed genes between RNAseq datasets derived from two different conditions; for example datasets could be obtained from samples before and after treatment of a particular drug or it could be comparison between normal and diseased state. The tools used in this pipeline and their utilities are as follows:
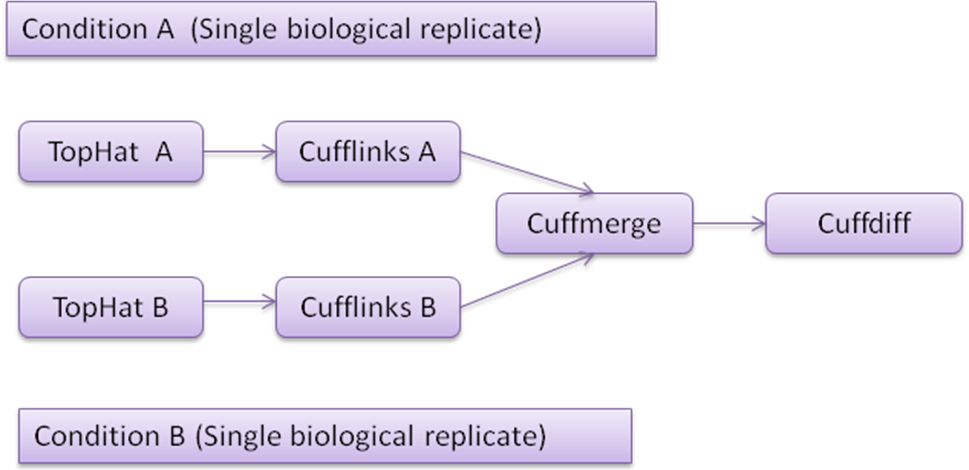
TopHat: TopHat is a splice junction mapper for RNAseq reads. It first aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie, and then analyzes the mapping results to identify exon-exon splice junctions.
Cufflinks: Cufflinks utilizes the information of aligned RNAseq reads (obtained from TopHat) and assembles the alignments into a parsimonious set of transcripts and calculates FPKM for each transcript.
Cuffmerge: Cuffmerge takes assembly from different Cufflinks output and merges them into a single unified transcript catalog.
Cuffdiff: Cuffdiff calculates significant changes in transcript expression, splicing, and promoter use.
Fig1: Typical RNAseq workflow for single replicate
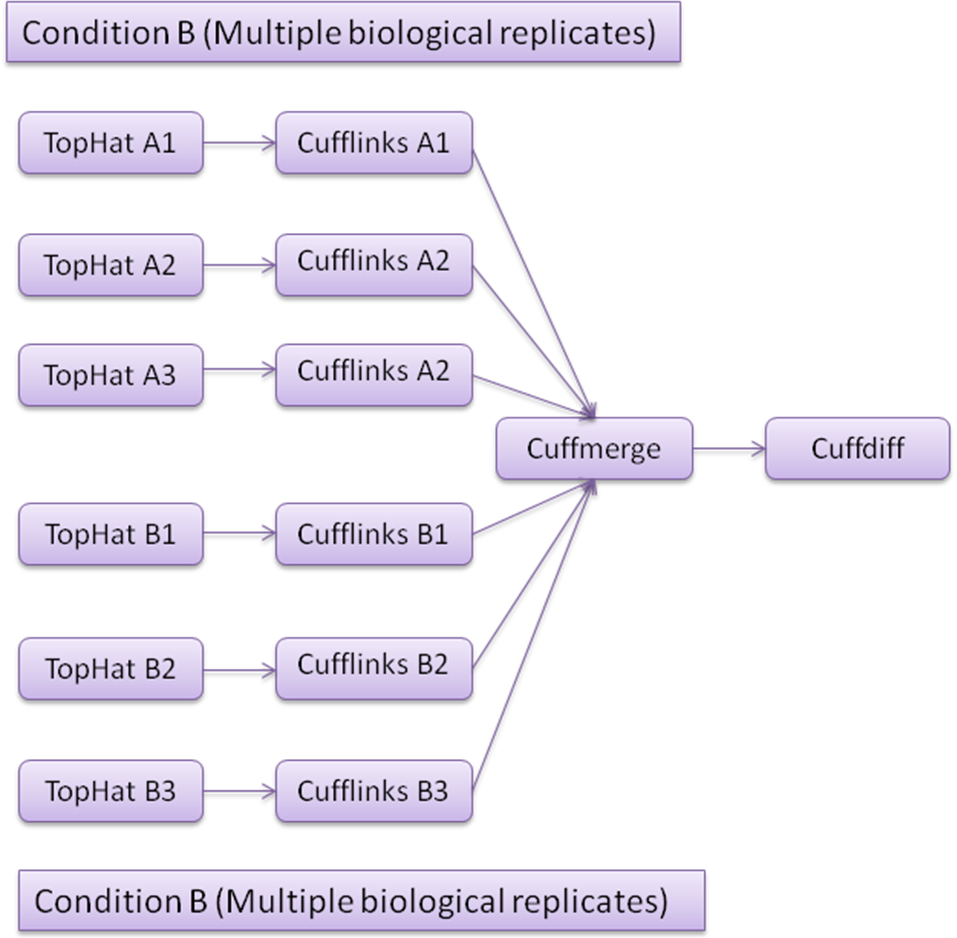
Fig2: RNAseq workflow for multiple replicates