Workflow for EST Assembly
Summary
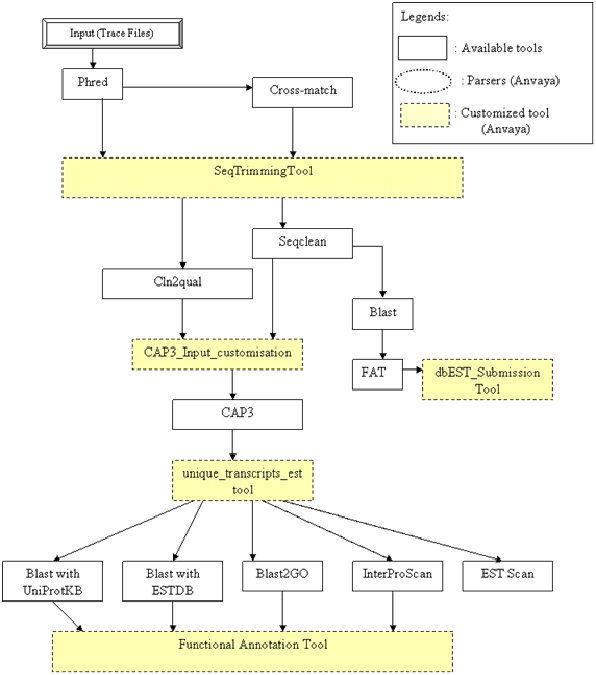
EST Assembly is one of the challenging workflow in bioinformatics research area. Here Anvaya provides researcher the single workflow, which will take the raw trace files from sequencing machines and will provide the fully annotated assembled ESTs.
The trace files from sequencing machines are processed by PHRED software for base calling. PHRED will provide the files containing sequences along with corresponding quality values. The sequences are further processed by the Cros_match tool for vector masking. The STT-Sequence trimming tool takes these vector masked EST sequences as an input. STT will remove the vector-masked nucleotides, polyA/T tail, polyC/G tail and Adapter/Linker sequences from EST sequences. STT will also maintain the similar changes/edition in the corresponding quality values. Further, STT-processed sequence files are processed by Seqclean for the removal contamination such as mitochondrial sequences. Seqclean will provide the filtered ESTs. The similar modification can be made in corresponding quality values by the tool cln2qual from seqclean package. One can remove the host/parasite contamination by the use of seqclean tool.
The filtered EST sequences thus obtained are further annotated by using BLAST. FAT tool will parse the BLAST results and will give EST sequences with their annotation in their header lines. The ‘dbEST submission files Tool’ takes these EST sequences along with their annotations as an input and provides the dbEST submission files having file format mentioned by NCBI.
The filtered EST sequences are submitted for assembly to Cap3 assembly tool. The ‘Unique Transcripts EST Tool’ combines the output files from Cap3 (assembled ESTs i.e. contigs file and unassembled ESTs i.e singlets file) and gives the single fasta formatted file containing the union of contigs and singlets sequences. This assembled EST dataset are further processed by BLAST, InterProScan, BLAST2GO for it’s functional annotation. The outputs from all these tools will be parsed by FAT-Functional Annotation tool for the fasta formatted sequences having annotation information in the respected header information. ESTScan will help in analyzing whether the sequenced ESTs are true ESTs or not.
Standard Tools
· Phred
· cross_match
· seqclean
· BLAST
· CAP3
· InterProScan
· ESTScan
· BLAST2GO
Customized Tools:
· SeqTrimmingTool
· unique_transcripts_est_tool
· dbEST_SubmissionTool
· FAT-FunctionalAnnotationTool
· CAP3_Input_customization
|
|
|
Fig:Translated implementation |
Details of workflow and tools used:
Ø Input files:
Directory location, which contains all the chromatogram files i.e. trace files.
Ø Base calling from trace files: Phred:
Phred processes input directory of trace files for base calling. Phred will predict fasta sequences and respective quality values per trace file.
Ø Cross_match: Vector masking:
A general-purpose utility (based on a "banded" version of SWAT, an efficient implementation of the Smith-Waterman algorithm) for comparing any two sets of (long or short) DNA sequences. For example, it can be used to compare a set of reads to a set of vector sequences and produce vector-masked versions of the reads.
Ø SeqTrimmingTool:
Sequence trimming tool will remove the vector-masked regions from the EST sequences. It also removes polyA/T tails, poly C/G tails, adaptor and linker sequences.
Input files:
1. The cross_match vector masked sequence output file (*.seq.screen file)
2. The Quality value file produced by Phred tool
Input parameters:
1. 5’ adapter sequence string
2. 3’ adapter sequence string
3. PolyA tail length (Same parameters will be set for polyT/C/G tails)
a. Should be greater than 0
b. Default value: 8
4. No. of mismatches allowed in polyA tail region (Same parameters will be set for polyT/C/G tails)
a. Should be greater than 0
b. Default value: 2
Output files: (need to be mention as an parameter)
1. Filtered Sequence file (*.seq)
2. Filtered Quality value file (*.qual)
3. Data filtering report file
Ø SeqClean and cln2qual:
A script for automated trimming and validation of ESTs or other DNA sequences by screening for various contaminants, low quality and low-complexity sequences. The cln2qual script will introduce the similar changes in quality value file as per the SeqClean report.
Ø dbEST_SubmissionTool:
This program is a part of Anvaya package, which creates the submission files for dbEST from the filtered and annotated EST sequences.
Input files:
1. File of EST sequences with their annotation (output file from FAT tool)
2. Laboratory information file having information like, Name of the research group, Contact address of research lab, organism details, primers details, vector information, Restriction enzyme details, publication details etc.
Output file (need to be mention as an parameter):
The output file containing records (EST sequence information) in NCBI dbEST submission format
Ø CAP3_Input_customization:
This is a customized tool provided as a part of Anvaya package. This will create the input file names compatible with the CAP3 software.
Input files:
1. Filtered sequence file from seqclean tool
2. Filtered qual file from cln2qual tool
3. Tag of output file
a. Default value: Cap3_In
Output files:
Sequence and qual file should be at the same location. User can not provide the name of output files. User can only provide the output tag. According to the output tag, the cap3 input files will be created.
1. Renamed sequence file i.e. <Tag>.seq
a. Default: Cap3_In.seq
2. Renamed quality value file i.e. <Tag>.seq.qual
a. Default: Cap3_In.seq.qual
Ø CAP3: Sequence assembly tool:
Cap3 is a sequence assembly program which process output files of CAP3_Input_customization tool. Cap3 will give 3 important files viz,
1. Assembled EST sequences i.e *.Contigs files
2. Unassembled EST sequences i.e *.singlets files
3. Cap3 system captured output where the user can find mapping of all EST with respect to contigs.
Ø Unique_transcripts_est_tool:
This is customized tool provided as a part of Anvaya package. This will give the concatenated file containing the all contigs and singlets.
Input files:
1. File containing contigs sequences. (Cap3 tool output)
2. File containing singlets sequences. (Cap3 tool output)
Output file name need to mention as an input parameter.
Ø BLAST Basic Local Alignment Search Tool (BLAST-2.2.14):
BLAST will search the unique transcript sequences against the available protein and dbEST databases. BLAST with dbEST will provide the information related to already available EST sequences in the dbEST database, which are similar to user’s EST data.
Ø InterProScan:
InterProScan is a tool that combines different protein signature recognition methods native to the InterPro member databases into one resource with look up of corresponding InterPro and GO annotation.
Ø BLAST2GO:
Blats2GO provides the Gene ontology terms for the query sequences. It takes the BLAST output in xml format as an input file.
Ø ESTScan:
ESTScan predicts the EST sequences. The prediction of this tool will help researchers to know how much accurate is the Query EST dataset.
Ø FAT-FunctionalAnnotationTool:
This is customized tool developed as a part of Anvaya package. It takes the outputs of different programs and parses them to give the fasta-formatted file having annotation details in the header location of each sequence.
Mandatory options:
1. Input file containing fasta-formatted dna/protein sequences
Optional input files:
1. Output file from BLAST program
i. Query Coverage cut-off [Default value: 80%]
2. Output file from InterProScan program (XML output)
3. Output file from BLAST2GO (*.annot file)