Workflow for Functional Annotation
Summary
Functional Annotation workflow template provides the easy way of annotating whole proteome set. This workflow uses multiple tools to annotate the given protein sequence. BLAST is used for functional annotation based on similarity with existing protein databases such as UniProt, nr etc. PfamHmm is used to identify the functional domains which are present within the sequence. Using programs viz, TMHMM and SignalP, user can predict subcellular localization. InterProScan is used to assign the Gene Ontology terms to the given protein sequences. The output of above mentioned programs is processed further by FAT-Functional Annotation Tool in order to provide protein sequences with annotations mentioned in respected header lines of fasta sequences.
|
|
|
Fig:Translated implementation |
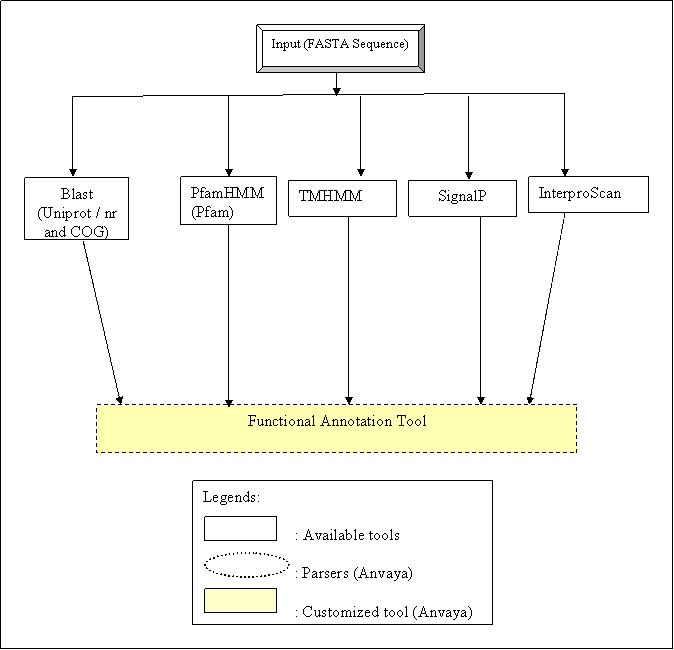
Standard Tools
BLAST, PfamHMM, TMHMM, SignalP, InterProScan
Custom tool
FAT- FunctionalAnnotationTool
Details of workflow and tools used:
Ø Input files
o File containing Fasta formatted protein sequences
Ø BLAST Basic Local Alignment Search Tool (BLAST-2.2.14):
BLAST will search the query protein sequences against the available protein databases.
Ø PfamHMM
PfamHMM searches the query sequence against the available sequeneces from Pfam to predict the domains in the given sequences.
Ø TMHMM
TMHMM is used in for prediction of transmembrane helices in proteins.
Ø SignalP
SignalP predicts the presence and location of signal peptide cleavage sites in amino acid sequences from different organisms: Gram-positive prokaryotes, Gram-negative prokaryotes, and eukaryotes.
Ø InterProScan
InterProScan is a tool that combines different protein signature recognition methods native to the InterPro member databases into one resource with look up of corresponding InterPro and GO annotation.
Ø FAT-FunctionalAnnotationTool
It takes the outputs of different programs and parses them to give the fasta-formatted file having annotation details in the header location of each sequence.
Mandatory options:
1. Input file containing fasta-formatted protein sequences
Optional input files:
1. Output file from BLASTP program
i. Identity cutoff [Default value: 90%]
ii. Query Coverage cut-off [Default value: 80%]
2. Output file from PfamHMM program
3. Output file from TMHMM program
4. Output file from SignalP program
5. Output file from InterProScan program (XML output)