Workflow for Genome Annotation
Summary
Genome annotation workflow is used to annotate a newly sequenced genome. This workflow can be used for both prokaryotic and Eukaryotic organisms.
Glimmer/Genscan predicts the genes from a given input genome sequence. In case of prokaryotes, glimmer results are further accompanied by ribosomal binding site prediction from RBSfinder. The tRNA genes are predicted by tRNAScan-SE program. The repeats from the genome are masked by using RepeatMasker program. All the results from Glimmer/Genscan, RBSfinder, tRNAScan, and RepeatMasker programs are combined to give in a standardized format i.e., in Genbank format. The client can view this genbank formatted result file using Artemis visualization tool for detailed analysis.
The Compseq utility from EMBOSS package is used for reporting composition of dimer/trimer/etc words in a sequence. The freak utility from EMBOSS package is used for providing residue/base frequency tables or plots. The restrict utility from EMBOSS package is used to find restriction enzyme cleavage sites within the given genome
|
Fig:Translated implementation |
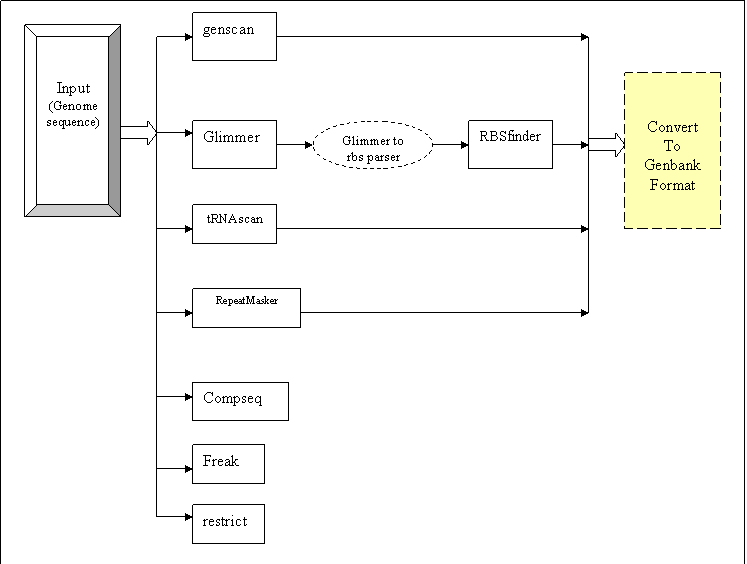
Standard Tools
Glimmer, Genscan, RBSFinder, tRNAScan, RepeatMasker, EMBOSS (compseq, freak, restrict)
Custom Tools
CGF: ConvertToGenbank Format
Parsers:
Glimmer_to_rbsfinder
Details of workflow and tools used:
Ø Input files:
o Prokaryotic Genome (Prokaryotic Genome annotation)
o Eukaryotic Genome (Eukaryotic Genome annotation)
Ø Gene prediction
o Glimmer (3.02)
Glimmer is a microbial gene prediction tool, which will predict the genes from given input genome. There are two options in order to predict the genes from a given genome.
1. Input_Type_Genome:
The ‘extract’ program will provide the ‘long-orf’ dataset from a provided genome sequence to ‘build-icm’ program in order to build the icm model. This icm model will be further used by ‘glimmer’ program to predict the genes from a given input genome.
2. Input_Type_multifasta:
The ‘build-icm’ program will use the provided multifasta gene dataset file to build the icm model. This multifasta gene dataset file may be from different organism as compared to organism of given genome sequence. This is an extra input file which user needs to upload at I/O node at the start of the workflow. The icm model thus obtained is further used to predict the genes of the given input genome sequence.
o RBSFinder
In case of prokaryotic genome annotation, the glimmer output is further processed by RBSfinder to predict the ribosomal binding sites.
One need to use the parser ‘glimmer_to_rbsfinder’ for providing the glimmer output (i.e *.predict file) to RBSFinder program. The parser will remove the first header line, which starts with the symbol “>” from glimmer output file.
o GenScan
GENSCAN is a general-purpose gene identification program, which analyzes genomic DNA sequences from a variety of organisms including
human, other vertebrates, invertebrates and plants.
Ø tRNAScan
tRNAscan-SE identifies transfer RNA genes in genomic DNA or RNA
Sequences.
Ø RepeatMasker
RepeatMasker is a program that screens DNA sequences for interspersed
repeats and low complexity DNA sequences.
Following are the outputfiles:
1. *.masked file: contains query sequence with all repeats and low complexity regions masked.
2. .out file: list of masked positions in the sequence and their annotation.
3. .tbl file: summary of the repeat content of the analyzed sequence.
Ø EMBOSS package programs viz, compseq, freak and restrict will provide the genome statistics
Ø CGF - Convert to Genbank format custom tool
This program will take the input from different nodes and will summarize the results in genbank format.
Mandatory options:
1. Input Genome sequence file
2. Glimmer/Genscan prediction output file
Optional input files:
1. RBSFinder output file
2. tRNAScan output file
3. RepeatMasker output file (*.out)