Workflow for promoter
identification using Microarray data
Summary:
The
workflow helps us to identify consensus patterns in the upstream regions of
certain genes, which may be clustered together, which may also lead to the identification
of novel target genes for therapeutic purposes. Gene expression data is given
as input for the workflow, for which the data is supposed to be pre-normalized
at the user’s end. The normalized data is then parsed for Cluster analysis
(preferably K-Means or hierarchical Clustering). For the different clusters
obtained, the corresponding gene ids are obtained from database followed by
retrieval of the upstream regions of the gene. Motif analysis tool MEME is used
to identify conserved patterns/motifs. The motifs thus obtained are searched
against the desired motif databases using the tool MAST.
Input
files required for this workflow:
- Gene expression data in .pcl
format with the first column or UID column having the standard NCBI
accession id, or locus tag/synonym or PID (protein ID).
- The desired database to be
searched (in fasta format) with the reported motifs by MAST tool.
|
|
|
Fig:Translated implementation |
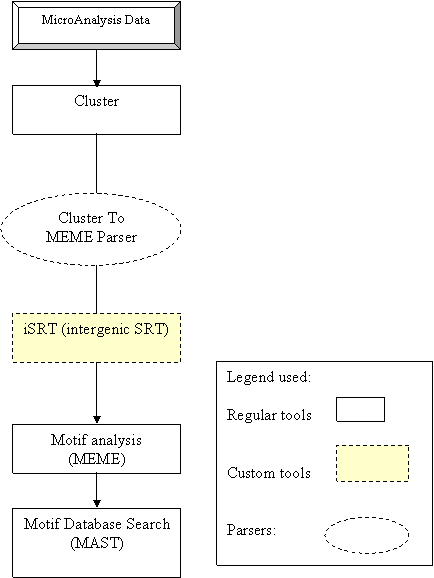
Standard Tools:
CLUSTER:
Perform
a variety of types of cluster analysis and other types of processing on large
microarray datasets. Currently includes hierarchical clustering, k-means
clustering, etc.
MEME:
MEME is
a tool for discovering motifs in a group of related DNA or protein sequences.
MAST:
MAST is a tool for searching
biological sequence databases for sequences that contain one or more of a group
of known motifs.
Custom
Tools:
- iSRT (intergenic Sequence
Retrieval Tool)
It is a custom tool that retrieves the
upstream (intergenic) regions of genes, given the gene ids, from the database
of intergenic regions. It takes as input a list of gene ids (synonym OR
protein_id OR PID), minimum length of the retrieved sequences, the sequence
start site and the sequence end site as arguments and retrieves the desired
sequences from the intergenic database.
Usage of this tool:
./intSeqRetreiver.pl id_file_name(inputFile) column_name minimum_req_length
required_length_of_db_sequence startPosition endPosition output_file_name
Where,
- intSeqRetreiver.pl is the tool
- d_file_name is the file which
contains the list of synonyms OR protein_ids OR PIDs for which the sequence is
to be retrieved in FASTA format (with complete path);
- column_name for unique
identification of an organism (whether synonym OR protein_id OR PID)
- minimum_req_length is
the minimum length of retrieved sequences (for eg, a sequence containing
atleast 50 nucleotides should be considered)
- required_length_of_db_sequence
is the maximum length of the sequence required
- Seqstart is the start
position from where the sequence is to be retrieved in the database
- Outfile is the name of
the output file in which the output is to be written.
Output of this tool:
1.
A multi-fasta formatted text file containing the intergenic sequences
Parsers:
- Cluster to MEME Parser
This parser gives a textual output of
the list of gene ids according to the clusters formed. According to the
co-relation cut-offs mentioned, the parser clubs the set of genes which fall in
the same cluster. According to the gene of interest provided by the user, the
cluster containing the gene of interest is passed on to iSRT (intergenic
Sequence Retrieval Tool) and upstream sequences of the gene ids are retrieved
on which MEME is run.