Workflow for Motif
Identification
Summary:
The
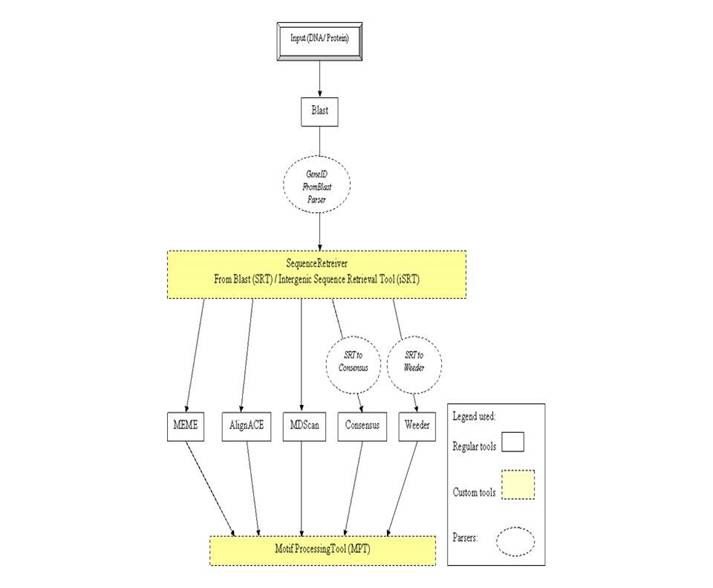
workflow identifies conserved patterns or motifs using DNA or Protein sequences
as input. The input sequences are searched against a preferred database using
BLAST and the significant hits obtained (orthologs) are parsed for the next
step of sequence retrieval. Motif discovery programs like MEME, AlignACE,
MDScan, Weeder and Consensus are used for pattern identification. The conserved
regions obtained are then parsed and analyzed through a custom tool “Motif
Processing Tool”.
Input
files required for this workflow:
- Protein/DNA
sequence of the query for which motif identification is to be done
Standard Tools:
·
BLAST
Find
regions of similarity between biological sequences.
MEME:
MEME
is a tool for discovering motifs in a group of related DNA or protein
sequences.
·
AlignACE
AlignACE
(Aligns Nucleic Acid Conserved Elements) is a program which finds sequence
elements conserved in a set of DNA sequences. It uses a Gibbs sampling strategy
·
MDScan
A DNA
sequence motif finding program.
·
Consensus
This program
determines consensus patterns in unaligned sequences.
·
Weeder
Finds
"Motif (transcription factor binding sites) discovery in sequences from coregulated
genes of a single species.
Parsers
- GeneIDFromBLAST
Parser:
This
parser collects gene ids as per the required cut-offs mentioned by the user
from the BLAST output and passes onto Sequence Retrieval Tool or intergenic
Sequence Retrieval Tool, for which sequences are retrieved and reported in
Fasta format.
- SRT
to MDScan:
This
parser converts the fasta format file derived from SRT/iSRT to MDScan format.
- SRT
to Consensus:
This
parser converts the fasta format file obtained from SRT/iSRT to Consensus
format
Custom
Tools
- Motif
Processing Tool:
It
is a custom tool which converts outputs from different motif prediction tools
into a text-based visual format (for easy comparison of results). It requires
output of at least two tools to compare. By default it takes first five motifs
predicted by each tool. If predictions are less than five, then it takes all
predicted motifs into consideration. It takes as input the outputs of different
motif prediction programs and processes them to give the modified
easy-to-interpret formatted output.
Usage
of this tool: ./MPT -nmotifs <no_of_motifs(Max 5)> -i
<Seq_input_file> -[m|meme] <meme.txt> -[a|alignace]
<alignace.txt> -[c|consensus]
consensus.txt>
-[s|mdscan] <MDScan.txt> -[w|weeder] <weeder.txt> -o mpt.out
Where,
-MPT is the
tool
-Nmotifs is
the number of motifs to be considered from each to be reported; the maximum
number of motifs to be considered is 5.
-Seq_input_file
is the file containing the sequences onto which the results are to be mapped
-Meme.txt is
the file containing the meme output
-Alignace.txt
is the file containing the alignace output
-Consensus.txt is the file containing
consensus output
-MDScan.txt is the file containing
MDScan output
-Weeder.txt is the file containing
weeder output
-Mpt.out is the output filename
Output of this tool:
1. A
text files containing the processed output of the motif prediction tools;
giving information about the sequences, and the positions where each tool has
predicted motifs to be present. Tabulated information of the same is also
provided.