Workflow for Ortholog
Prediction
Summary:
The
workflow predicts orthologs given a set of two genomes using the criteria of
bi-directional best hits. BLAST is used for detecting the orthologs. Initially
the gene sets of two organisms are subjected to BLAST and orthologs are
predicted in cases where the criteria of bi-directional best hits are
satisfied. Genes are referred to as unique if they do not satisfy the criteria
of bi-directional best hits. Such unique genes of a given organism are then
searched against the genome of the other organism so as to cross-check if the
genes have been missed due to the limitation of the gene prediction program. If
significant matches are found at the genome level then it is not a unique gene
and if no matches are detected then the genes are unique with respect to the
other organism in consideration.
Input
files required for this workflow:
- Proteome sequences in multi
fasta format of the two organisms for which orthologs are to be detected
(*.faa files)
Whole
genome sequences of both organisms (*.fna files)
|
|
|
Fig:Translated implementation |
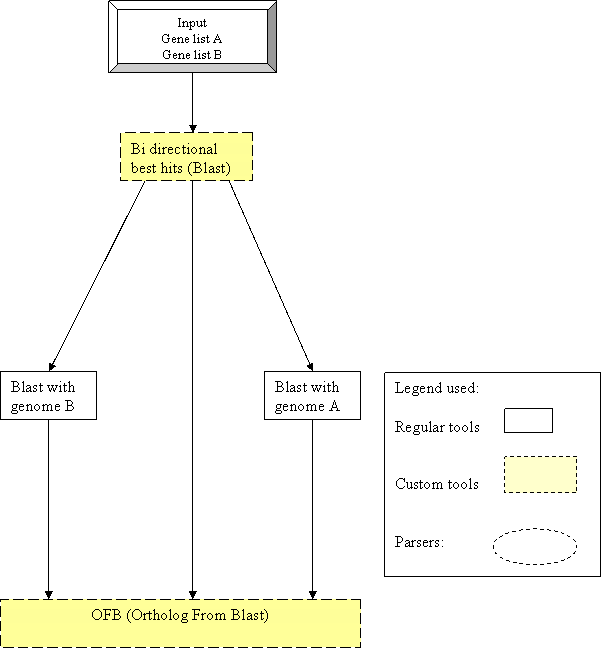
Standard Tools
·
BLAST: Find regions of similarity between biological sequences.
Custom tools
- Bidirectional Best Hit (BBH)
It is a custom tool which performs two
BLASTP programs at a time taking proteome of one of the organisms as input file
and proteome of the other as database and vice-versa. It also fishes out the
orthologs found using the bi-directional best hit criteria between the two
organisms.
Usage
of this tool: ./bbh.sh input_seq1 input_seq2 Output_Directory
Where,
bbh.sh is the tool
Input_seq1 and input_seq2 are the
proteome sequence files of two organisms respectively. (in multi fasta format)
Output_Directory is the directory path
for the output files.
Output
of this tool:
1.
orthologsQuery.out is the list of orthologs reported by BBH tool
2.
hitReject.out is the file containing list of genes from input_seq1 for which no
orthologs were reported. This goes as an input to TBLASTN in the next step.
3.
queryReject.out is the file containing list of genes from input_seq2 for which
no orthologs were reported. This also goes as an input to TBLASTN in the next
step.
4.
out1.out and out2.out are files containing summary reports of BLAST runs.
Summarized BLAST report of input_seq1as query and input_seq2 as database is
reported in out1.out and summarized BLAST report of input_seq2 as query and
input_seq1 as database is reported in out2.out
- Ortholog from BLAST (OFB)
It is a custom tool which reports the
final orthologs, if found from two TBLASTN output, given the desired identity
and query coverage as well as unique genes found in the respective two genomes.
It takes as input three files, orthologsQuery.out file from BBH, and the two
TBLASTN output from the previous nodes. Given the desired identity value and
percentage query coverage, it looks for genuine orthologs, if missed
previously, and appends the result to the file orthologsQuery.out. The genes
which did not satisfy the given identity and percent query coverage criteria
are reported as unique genes.
Usage of this tool: ./ofb.pl
first_input_file_name second_input_file_name orthologsQue Identities
Query_Coverage output_directory
Where, ofb.pl is the tool
First_input_file_name is the first
TBLASTN output (i.e., TBLASTN result of queryReject.out as query with GenomeB
as database)
Second_input_file_name is the second
TBLASTN output (i.e., TBLASTN result of hitReject.out as query with GenomeA as
database)
orthologsQue is the orthologsQuery.out
file reported by BBH
Identity is similar to BLAST identity
values and this parameter takes integers as input
Query_coverage is the percentage query
coverage, which tells us how well our query is aligned to the database
provided. It is calculated as follows:
% query coverage = ({(query end – query start) + 1}/ query length)*100
Output of this tool:
1.
orthologsQuery.out is the appended list of orthologs reported by OFB
2.
Uniquegene_A is the list of genes present in genomeA (first genome) for which
no orthologs were reported against genomeB (second genome)
3.
Uniquegene_B is the list of genes present in genomeB (second genome) for which
no orthologs were reported against genomeA (first genome).
4. temp_out
is a file containing summarized reports of the two TBLASTN runs.