Workflow for
‘Phylogenetic profiling to detect functional linked genes’
Summary
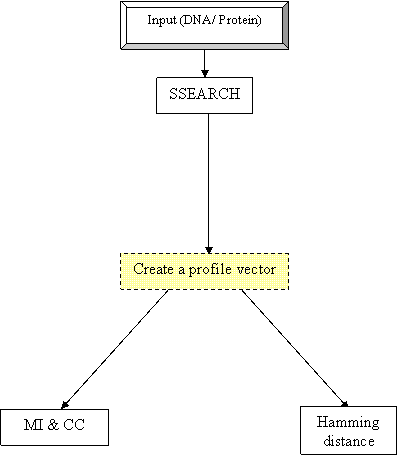
The workflow
aims to infer functional linkages using phylogenetic profiling. The input
protein sequence(s) is usually from any organism with its complete genome
sequenced which is searched against proteome data of other organisms with
completely sequenced genome using either BLASTP/SSEARCH. The e-values obtained
after search of every protein sequence is parsed and normalised and represented
as a profile vector/matrix where in the rows are individual proteins and the
columns are organisms. The profiles obtained are analysed for their statistical
significance using parameters like mutual information content, hamming distance
and correlation coefficient.
|
|
|
Fig: Translated implementation |
Standard Tools:
·
SSEARCH:
Implements MPI version of Smith-Waterman algorithm for database searching and
reports the alignment along with statistical significance of the hits obtained.
·
mis_calc:
Calculates mutual information content and Pearson Correlation Coefficient of a
given profile pair.
·
Custom
Tools:
- Create_profile_vector:
Reads in the results of independent
Smith-Waterman database searches and creates a matrix containing normalized
E-values.
The SSEARCH output is parsed in terms
of % query overlap (default: 40%). The e-value is normalized using the formula
normalized e-value=-1/log E
The tool reads in outputs of independent
search (one per organism) and creates a matrix with normalized E-values. The
following are some of the assumptions made for the construction of the matrix:
5 --> refers to No significant Hits
Found query.
1 --> refers to eVal >= 1
0 --> refers to eVal = 0
2 --> refers to eVal >0 and
<1 BUT NOT satisfying conditions.
normaliseEval --> refers to
eVal > 0 and < 1 satisfying conditions.