Workflow for Phylogeny
Summary
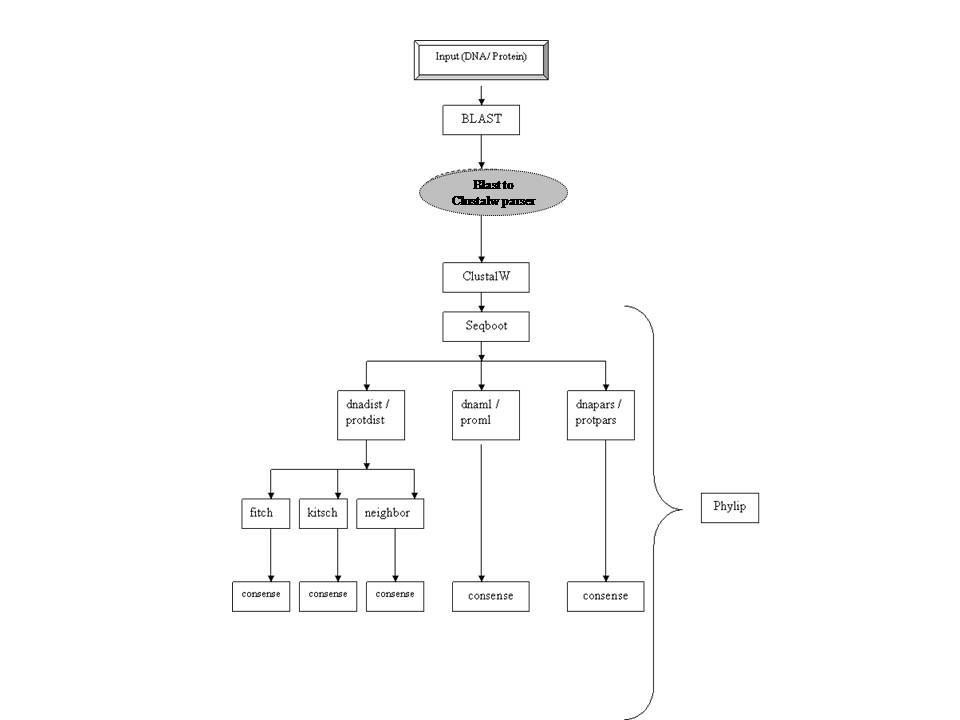
The
workflow builds a phylogenetic tree of the orthologs detected for a given query
sequence (nucleotide or protein) using a similarity search tool. User needs to
provide only the query sequence to carry out a similarity search using BLAST
against a chosen database. Parsers will redirect the output of BLAST to
multiple sequence alignment tool, ClustalW, which would then pass the output to
Phylip suite for reconstruction of phylogenetic tree of the orthologs.
|
|
|
Fig:Implemented Translation |
Parser in this workflow:
BLAST to ClustalW
o
Reads
the results of BLAST search and creates a multi-fasta file containing sequences
that satisfy user-defined parameters of E-value, % identity, % overlap and bit
score.
o
Default values of the parameters are:
·
E-value:
0
·
%
identity: >= 30
·
%
query coverage: >=50
·
%
database hit coverage: >=50
·
Bit
score: >=40
·
Sequence Retrieval Tool: Retrieves the fasta sequences of the entries
satisfying the above mentioned criteria from the corresponding table of the
database stored in a RDBMS like MySQL. The storage of standard databases like
UniProt and nr as tables in MySQL increased the pace of retrieval which
otherwise had to be extracted from a text file with upto 3GB size and consumed
lot of time for the same task.
Standard Tools :
·
BLAST: BLASTP and BLASTN are used for
detection of orthologs for the query sequence. The BLAST output is parsed in
terms of e-value, % query overlap, % sequence identity and bit score.
·
ClustalW: The MPI version of ClustalW is used
for multiple sequence alignment of the orthologs detected using BLAST. The MSA
output is converted into Phylip format using output format options of ClustalW.
·
Phylip
suite: Is used for
reconstruction of phylogenetic trees. Multiple datasets are generated using the
‘seqboot’ program. Different methods and the corresponding programs used for
tree building are:
o
Distance
based: dnadist, protdist, fitch, kitch, neighbor
o
Parimony
based: dnapars, protpars
o
Maximum
Likelihood based: dnaml, proml
A final majority consensus
tree is built using ‘consense’ program. The trees can be viewed by installation
of visualizer like ‘TreeView’ on the client machine.