Workflow for Prediction
of Antigenic sites
·
Summary
The
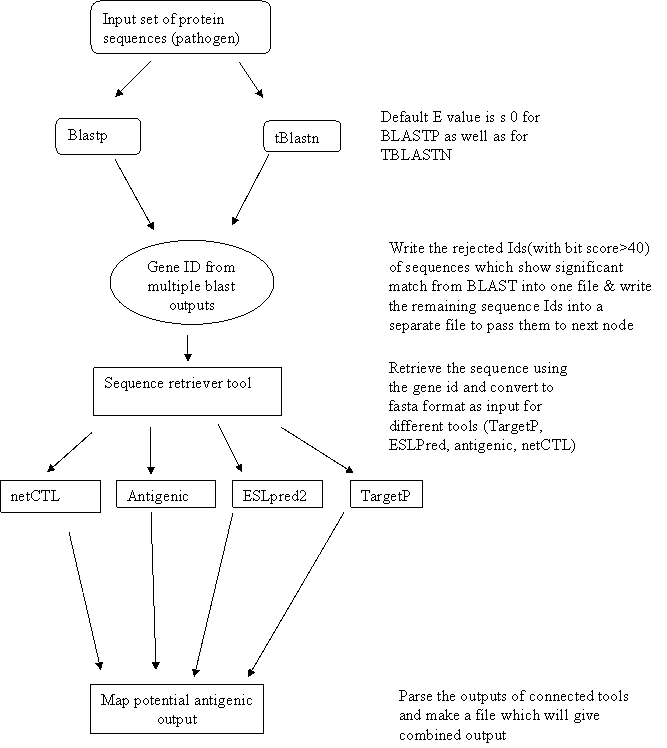
workflow is designed to predict the potential antigenic regions in a given set
of proteins of a pathogenic organism. In this workflow multiple BLAST programs (BLASTP,
TBLASTN) are run for queried Pathogen sequences against the host sequence (nr
for BLASTP and human/mouse for TBLASTN) databases. The E value for BLAST
searches is 0. A parser is also built to write the rejected Ids of sequences
which show significant hits from BLAST into one file and write the remaining
sequence Ids into a separate file to pass them to next node (Take the geneid as
input from BLASTP/TBLASTN and output is used as input to SRT).
The
short listed sequences have BLAST bit score <= to 40 and query coverage
>= 50 as default. Sequences having low score or no match are the unique
pathogen sequences. These sequences are retrieved from the BLAST output by
using GeneID from multiple BLAST parsers and saved in flat file format
comprising of fasta sequences using in-house built Sequence Retrieval Tool.
Selected
sequences are than used as input for tools like netCTL, Antigenic, TargetP and
ESLpred2 to predict T cell and B cell epitopes along with their sub cellular
location.
netCTL
predicts the cytotoxic T cell lymphocyte epitopes. netCTL has been chosen
because of its higher predictive performance than EpiJen, MAPPP, MHC-pathway,
and WAPP on all performance measures. T cell epitopes are predicted and stored
in flat file format using custom tool ‘Map potential antigenic output’
Antigenic
(emboss) is used to predict B cell epitopes. The probable B-cell epitopes are
predicted and retrieved by using Map potential antigenic output custom tool.
TargetP
and ESLpred2 are used to predict the sub cellular localization of the given
sequences and if the sequence is detected as secretory or extra cellular
protein or contains signal peptides more weight is added to
the netCTL and antigenic results.
·
Standard Tools
- BLAST
- NetCTL
- Antigenic
- ESLPred2
- TargetP
·
Custom Tools
- Sequence
Retrieval Tool
- Map
Potential Antigenic Output
·
Parser
·
GeneID from Multiple BLAST Outputs
|
|
|
Fig: Translated implementation |
·
netCTL parameters (red color text are the
parameter used)
a. Weight on C
terminal cleavage: The default value (0.15) gives
optimal predictive performance on average. The user can modify the relative
weight on proteasomal cleavage by entering a different weight value.
b. Weight on TAP
transport efficiency: The default value (0.05)
gives optimal predictive performance on average. The user can modify the
relative weight on TAP transport by entering a different weight value.
c. Threshold for
epitope identification: Peptides with a combined prediction score value greater
than the threshold value are marked as potential epitope. In a large scale
benchmark identifying known CTL epitope in proteins the default value of 0.75 was found to correspond to a sensitivity of 0.65
and specificity 0.97.
d.
12 class I binding MHC supertypes used: A1, A2,
A3, A24, A26, B7, B8, B27, B39, B44, B58, B62
·
TargetP parameters:
Select the
organism non-plant, Perform cleavage site
predictions
Specificity
>0.95 (predefined set of cutoffs that yielded
this specificity on the TargetP test sets, specificity >0.90 (predefined set of cutoffs that yielded this
specificity on the TargetP test sets), Define your own cutoffs (0.00 - 1.00).
·
ESLpred2 parameters:
Specify method as 3
(Hybrid approach) is used to predict the sub cellular localization; select the
organism G (generalized dataset).
1 for AAC with terminals; 2 for PSSM Composition; 3 Hybrid
A for Animal dataset; F for Fungi; P for Plants dataset; G for Generalized
dataset
·
Antigenic parameter:
The
minimum length of antigenic region is 6 but can
be taken from 1-50.
·
BLAST parameters:
E value is 0
for BLASTP as well as for TBLASTN and the sequence having BLAST score <= 40 and query coverage >= 50
as default are taken for next node.